1. Prediction of protein-ligand binding using enhanced sampling methods
Molecular recognition is one of the central issues in biological science and in drug discovery. The current understanding largely relies on the classical “lock and key” model and on X-ray crystal structures. On the other hand, updated mechanisms that incorporate structural flexibility of proteins and ligands have been proposed and verified theoretically and experimentally. In addition, current strategy for designing drug compounds considers not only binding affinity to a target protein but also residence times. Molecular dynamics (MD) simulation, which explicitly incorporates solvent effects and protein structural flexibility, is a powerful tool for predicting protein ligand binding and association processes. However, in conventional MD simulations, it is difficult to directly observe ligand binding and protein association that occur on milliseconds time scale or longer. To overcome this difficulty, we develop free energy calculation methods based on a various enhanced sampling methods, such as the Replica-Exchange MD method (REMD method), and apply them to elucidate the dynamical mechanisms of molecular recognition.
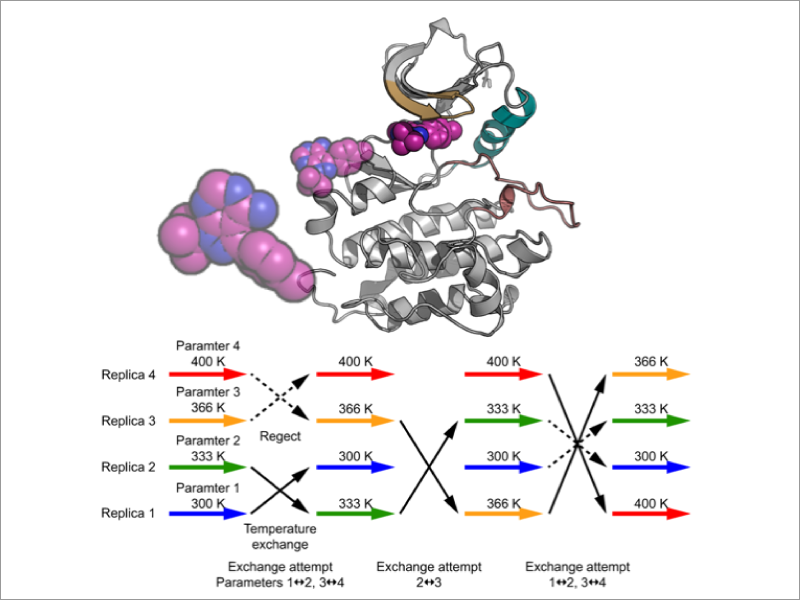
References
- Replica-exchange umbrella sampling combined with Gaussian accelerated molecular dynamics for free-energy calculation of biomolecules. Hiraku Oshima, Suyong Re, and Yuji Sugita. J. Chem. Theory Comput., (2019) DOI:https://doi.org/10.1021/acs.jctc.9b00761.
- De Novo Prediction of Binders and Nonbinders for T4 Lysozyme by gREST Simulations. Ai Niitsu, Suyong Re, Hiraku Oshima, Motoshi Kamiya, Yuji Sugita. J. Chem. Inf. Model., 59, 3879-3888 (2019).
- Encounter complexes and hidden poses of kinase-inhibitor binding on the free-energy landscape. Suyong Re, Hiraku Oshima, Kento Kasahara, Motoshi Kamiya, and Yuji Sugita. Proc. Natl. Acad. Sci. USA, 116, 18404-18409 (2019).
- Flexible selection of the solute region in replica exchange with solute tempering: Application to protein-folding simulations. Motoshi Kamiya and Yuji Sugita. J. Chem. Phys., 149, 072304 (2018).
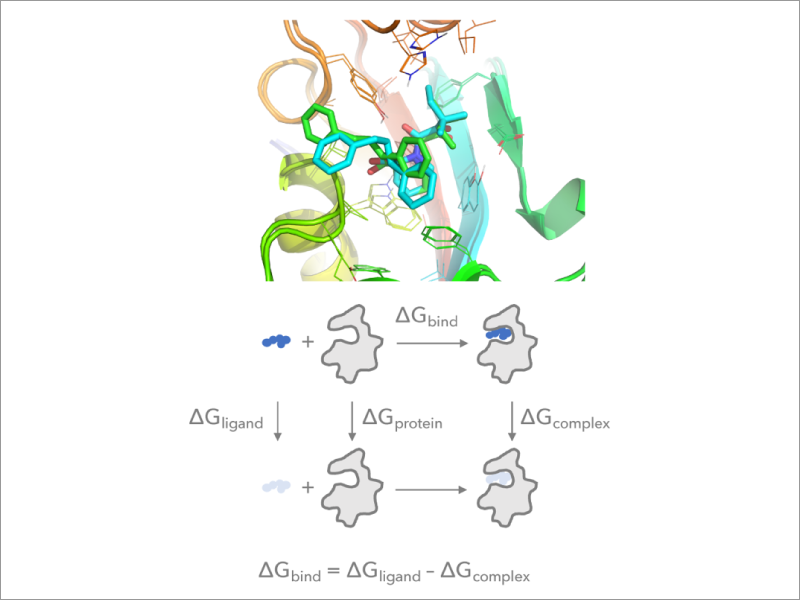
2. Implementation of the free energy perturbation method in GENESIS
Free Energy Perturbation (FEP) calculation using MD simulations is the most commonly used approach to predict free energy changes (both absolute and relative changes) accompanying protein-ligand and protein-protein association. FEP calculations are computationally expensive because the free energy change between two states is evaluated by introducing many intermediate states. We develop an efficient FEP method which takes full advantage of massively parallel supercomputers and implement it to GENESIS. We further combine the method with various enhanced sampling techniques in order to predict free energy changes of different biomolecular processes with high precision.
Prediction of protein-ligand binding using enhanced sampling methods
Implementation of the free energy perturbation method in GENESIS